This section describes the rules for encoding of carbohydrate and derivative structures in a single line ('CSDB linear code'). You may need this information in four cases:
Content:
Residues are described by a sequence of terms like <residue name>(<linkage>). In the case of the reducing end residue, the expression in parentheses is not required. For example, A(1-3)B(1-4)C is a linear fragment, in which residue A substitutes position 3 of residue B by its first position, residue B substitutes position 4 of residue C by its first position, and residue C substitutes nothing. Here and below, Latin capitals stand for residue names.
|
|
The logic of residue connection is a tree graph. The rightmost residue in an oligomeric structure (reducing end) is its root. Every residue except root must substitute a single residue. Any residue, including root, can have zero or more substituents. Substituent is a residue at the left (e.g. glycosidic bond donor, or a child in a tree graph); substituted residue is one at the right (e.g. glycosidic bond acceptor, or a parent in a tree graph). Polymeric structures have a pseudo-root, which substitutes another instance of the same repeating unit, thus forming a cyclic graph.
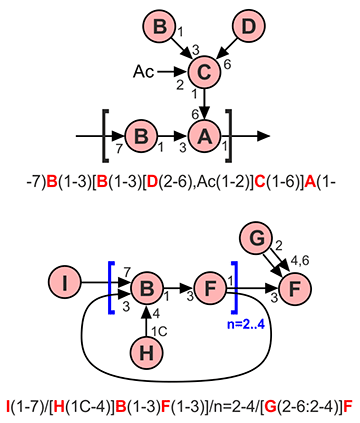
If there are branching points, one chain is always considered as main, and others are side chains. To distinguish which chains are main and which are side, refer to the corresponding section below. The side chains are enclosed in square brackets together with parentheses indicating a linkage. For example, ^^A(1-3)[B(1-4)]C is a branched fragment in which residue A substitutes position 3 of residue C, while residue B substitutes position 4 of residue C. In substructure search requests, the ^^ prefix indicates that the marked residue is unsubstituted. Several side chains attached to one residue are separated by commas. Side chains can also be linear or branched, and all combinations of nesting square brackets are allowed, e.g. -7)B(1-3)[D(1-3)[E(2-6)]C(1-6),G(2-6:2-4)F(1-4)]A(1-.
The linkage between residues is specified in parentheses to the right of the donor residue (<goes by>-<goes to>), where goes by denotes the position (carbon number) by which this residue substitutes another residue (usually 1 or 2), and goes to denotes which position of the linked residue is substituted. For example, 1-O-methyl,2-O-α-D-manno-β-D-glucoside is represented by aDMan(1-2)bDGlcp(1-1)Me.
Outgoing (goes by) and ingoing (goes to) linkage positions can be numbers or question marks (?) if unknown. The ingoing linkage position to Subst(N) aliases can contain position modifiers (a,b,c,',").
It is assumed that any linkage is formed with elimination of water or ammonia, giving ester (including glycosidic and phosphodiester), ether, amide or amine bond. To specify a carbon-carbon linkage, use C-character after the outgoing position (e.g. bD1dGlcp(1C-4)Subst // Subst = 2-aminopyridine = SMILES N{2}c1c{4}cccn1). Please note, that since there is no OH group at C1 of a sugar residue in C-glycosides, it is encoded as 1-deoxy-monosaccharide (1dGlc). To specify carbon-nitrogen linkage in N-glycosides and N-linked glycoproteins, please use 1N derivatives of monosaccharides to keep consistency with NMR simulation modules (e.g. bDGlc1N(1-4)xLAsn).
If a residue forms more than one outgoing linkages to its acceptor residue (pyruvates, biphosphates etc.), use colon to separate the linkages inside parenthesis, e.g. xRPyr(2-6:2-4)aDGal means the 4,6-pyruvated galactose. The higher position in acceptor always goes first (A(2-6:2-4)B but not A(2-4:2-6)B). Biphosphates and bisulphates have 0 as their <goes by> index, e.g. P(0-6:0-4)bDGalp.
Except for bisubstitution, phosphates and sulphates should be included into the linkage parenthesis like this: aDGlc(1-P-4)bLFuc (in the chain) or P-4)bLFuc (at the non-reducing end) or aDGlc(1-P (at the reducing end). Longer chains (di-, tri- etc.) are allowed: aDGlc(1-P-P-4)bLFuc or S-S-4)bLFuc
Although linkages are undirected, it is strongly recommended to follow left-to-right logic of substitution to comply with a tree representation of carbohydrates. To do this, arrange residues in a way to make anomeric centers (or other default linkage atoms in non-carbohydrate residues) the outgoing linkage positions, e.g. to record a disaccharide built of two aldoses A and B use A(1-2)B but not B(2-1)A (and C(1-3)[A(1-2)]B but not C(1-3)B(2-1)A). In some special cases (e.g. linkage between two anomeric centers: aDGlcp(1-2)bDFruf) it is impossible to apply this rule. In this case, minimize the numer of inverted linkages in a structure.
If a linkage produces another stereo-center, which was achiral in a free residue, and its configuration cannot be encoded as anomericity or absolute configuration (e.g. xSPyr for S-linked pyruvic acid diacetal), explain a newborn stereocenter as S or R after two slashes: xDGlca(1-1)Me // xDGlca = S-D-glucose (open). The same explanation can be used for carbon-carbon linkage producing a newborn stereo-center in Subst alias: bDGlcp(1C-2)Subst // Subst = 2S-1-aminopropane = SMILES C(N){2}CC (please note that 2S is not encoded in SMILES).
If a structure is reported as a repeating unit, the leftmost and rightmost residues should have open linkages, e.g. -2)A(1-3)B(1-4)C(1- means ABC fragment is repeated, and repeating units are linked to each other by 1-2 linkage. For polymerization frame positioning, see section Unit types and polymerization degree.
The repeating parts inside bigger structures can be described using the syntax A/B/n=?/C, where B is a repeated part of the structure, n=? is a repeat count (can be a number, range, or question mark if unknown), A is a capping part at non-reducing end, and C is a core part at reducing end. The whole structure can be polymeric as well (i.e. it can have open linkages at left and right), different repeating parts can be sequential or nested up to one level.
Examples:
Each residue name is composed of several fields following each other without separators:
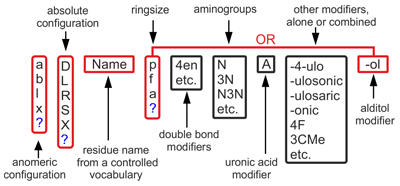 |
Monosaccharide base names are abbreviations of trivial names encoding (together with absolute configuration) stereochemistry of all chiral carbons. Below are some widespread examples:
Non-sugar base names are abbreviations of their trivial names:
Examples: aD6dTalA, aXKdo, xLGro, xDManN-ol, Ac, xRPyr, ?DFucN3N.
You can try to combine these fields and examine more names here.
Click here for the monomer namespace table
The naming of lipid residues matches the general naming described above. l should be used for anomeric configuration. For most lipids there are reserved names like Pam, Ole, Vac etc. (the complete list is available in the aliphatic acid section of the complete residue list). If there is no reserved base name, the following rules can be used to construct the new ones:
All monovalent substituents (Ac, Me, Et, Fo and other residues that CANNOT be substituted) should be described as separate residues, e.g. aDGal(1-3)bDGlcNAc should be recorded as aDGal(1-3)[Ac(1-2)]bDGlcN. If a monovalent residue is an aglycon at the reducing end, write it as following: aDGlc(1-1)Me. During the substructure search (but not during the data upload or automated exchange) you can specify monovalent residues in a common way, e.g. bDQuiNAc3NAc4Ac.
Phosphates and sulphates should be included into the linkage parentheses like this: aDGlc(1-P-4)bLFuc (in the chain) or P-4)bLFuc (at the non-reducing end) or aDGlc(1-P (at the reducing end). Longer chains (di-, tri- etc.) are allowed: aDGlc(1-P-P-4)bLFuc or S-S-4)bLFuc. However, in case of biphosphates or multiple substitutions, a regular syntax is allowed with 0 as a substitution position, e.g. NH2(1-0)P(0-2:0-3)bDGlcp
The chain is called normal if it is not secondary. The chain is secondary if it starts with any of the following residues (has it at the reducing end or consists only of it):
To keep this encoding unambiguous, you should pay attention to which chains are encoded as main and which are the side ones.
When comparing substitution positions, the following special cases exist:
A linkage is non-stoichiometric if its donating residue is present in a non-stoichiometric amount in polymer repeating units or the structure represents a mixture of oligomers. In this case, the residue name should be preceded by the stoichiometry degree in percents (e.g. 40%bDGlc). A single percent sign without a number (e.g. %Ac) means that the residue is present in a non-stoichiometric quantity but the exact amount is not known. Inline phosphate and sulphate residues can be preceded by percentage as well, implying non-stoichiometry of the bond between inorganic acid residue and its acceptor, e.g. xDRib-ol(1-50%P-4)bDGalp
The percentage is applied only to the outgoing linkage of the residue, which means that if a residue is substituted, all its children chains are non-stoichiometrical, too. For example:
The residue within a polymer backbone and the root residue in an oligomer cannot be non-stoichiometric. To encode structures like A-B-%C, use A[%C]B or %C[A]B.
Unknown anomeric or absolute configurations and ring sizes can be encoded at the residue name level (see above). For unknown linkage positions, use a question mark, e.g. Subst1(?-?)bLFucp or xXEtN(1-P-?)aXKdop. For residues that are not fully elucidated, see the Superclasses section.
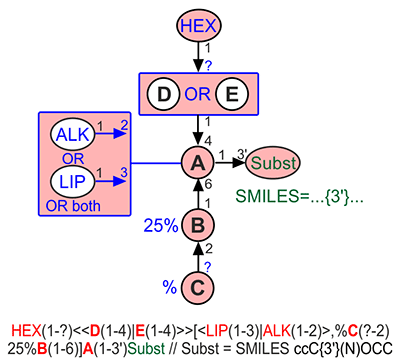 |
Fuzziness at the topological level can be encoded by one of two syntactic constructions: <<A(n-m)|B(p-q)>> for exclusive combination (XOR) and <A(n-m)|B(p-q)> for inclusive combination (OR). This means that e.g. <<A(1-3)|B(1-4)>>C is a disaccharide, in which either C3 of residue C is substituted by residue A or C4 of residue C is substituted by residue B, but not both at once, while <A(1-3)|B(1-4)>C is a disaccharide, in which C3 of residue C is substituted by residue A, or C4 of residue C is substituted by residue B, or both these positions are substituted by A and B, accordingly.
A fuzzy residue enclosed in angle brackets can be substituted itself, e.g. D(1-2)<<A(1-3)|B(1-4)>>C means that the structure is either D(1-2)A(1-3)C or D(1-2)B(1-4)C. If a variant inside a substituted fuzzy block is longer that one residue, it is assumed that the residue on its non-reducing end is substituted, i.e. A<BC|D>E is interpreted as ADE or ABCE, but not as ADE or A[B]CE. If a residue at the non-reducing end is monovalent or phosphate or sulfate, it is omitted from consideration and substitution focus is moved to its acceptor. If there are more than one non-monovalent non-reducing ends, the substitution focus is selected arbitrary. To reduce ambiguity and complexity of interpretation it is recommended to avoid substituted fuzzy residues and to add substitution to each of its variants in a fuzzy set (use E[<DA|DB>]C instead of E[D<A|B>]C). But if it is impossible, always use as short chains inside a fuzzy block as possible, i.e. -ED<A|B>C- but not -E<DA|DB>C-
If stoichiometry of branch reducing ends is specified inside a fuzzy block, e.g. -2)D(1-2)[<<E(1-4)30%A(1-3)|70%B(1-3)>>]C(1-, it is interpreted as ratio of variants. If the total sum of these values is less than 100%, or logic is OR, the whole fuzzy set can be missing. For example, <30%A(1-3)|70%B(1-4)>C means one of four variants (A(1-3)C, B(1-4)C, A(1-3)[B(1-4)]C, and C), while <<30%A(1-3)|70%B(1-4)>>C means one of two variants (A(1-3)C, B(1-4)C) and <<30%A(1-3)|60%B(1-4)>>C means one of three variants (A(1-3)C, B(1-4)C, and C). If a fuzzy set can become empty (variant C in previous examples), it cannot be substituted.
More than two variants inside a fuzzy block are allowed, e.g.<A|B|C>.
Angle brackets can be nested up to one level, e.g. <<D|<<A|B>>|C>> or <<<<A|B>>|C>> or <<<A|B>|C>> or <<<A|B>>|C> or <<C|<A|B>>> etc.
Possible risk: alternative sets are proposed mainly to specify a single variative residue. Although more sophicsticated features are supported, it is recommended to avoid them, as their interpretation can be ambiguous or misleading for chemists:
Unsupported:
If an exact residue at a certain position in the structure is unknown, a superclass name can be used instead of a residue name. Superclasses do not require anomeric and absolute configurations and ring size. The following superclasses are supported:
|
|
For rarely occuring sugars or non-carbohydrate residues or other residues that are not stored in the monomer database, use aliases. There are several allowed alias types:
All aliases other than Subst or SubstN should have anomeric and absolute configurations and ring size (which can be ?=unknown). All aliases should be explained in the comment section of the encoding after two slashes (//), e.g. aDGlc(1-4)Subst // Subst = Kdo-rich core, ID 12345. Several alias explanations are separated by semicolon, e.g. Subst1(?-6)aDGlc(1-1)Subst2 // Subst1 = acyl or polyglycerol; Subst2 = 3-hydroxy-2,4-diamino-toluene = SMILES ....
For fully defined aliases, add SMILES code after second equation mark if possible. Although trivial / IUPAC name can be omitted (e.g., Subst(1-6)aDGlcp // Subst = SMILES C=C{1}C(O)CO ), it is recommended to keep trivial or short systematic names for clarity.
Substitution positions should be indicated with a curly-bracketed atom number before the corresponding carbon: aDGlcp(1-3)Subst // Subst = laricytrin = SMILES O{3}C1=C(C2=CC(OC)={54}C(O){55}C(O)=C2)OC3=C({5}C(O)=C{7}C(O)=C3)C1=O. Details:
You can use online JSME structure editor to draw a structure and generate its SMILES code. To identify a substituted atom easily, mark a carbon of interest by -button and it will appear in square brackets with a number in the resulting SMILES. To generate SMILES, press . To check an existing SMILES code, use import feature (). Please keep in mind that you should draw complete residues including all -OH or -NH2 groups as they appear before water is eliminated upon linkage (e.g. draw methanol if you need to SMILESify an O-linked methyl group).
Greek letters, single and double quotes are allowed in Subst(N) explanations (ST1) and aglycons (AG). Substitution positions with single or double quotes or letters (3', 4", 15a) are allowed for Subst(N) residues only.
There is a special field AG to store aglycon information. Please, use it only if the aglycon cannot be encoded in the structure itself, i.e. the aglycon is a residue not supported by the database, or an non-encodable group of residues, or a superclass (see above). The aglycon attachment is assumed C1 for aldoses and C2 for ketoses. If possible, always try to encode a residue in structure rather than in aglycon:
| ST1: aDGalp(1-P-P-5)xXnucA AG: |
but not | ST1: aDGalp AG: ADP |
However, if the Subst or SubstN alias stands for aglycon (a moiety at the reducing end that cannot be encoded using the rules above) and it is attached to an unknown or default (C1 of aldoses, C2 of ketoses) position, it should be encoded in the aglycon (AG) field rather than in the structure (ST1), i.e.:
| ST1: aDGalp AG: lipid A |
but not | ST1: aDGalp(1-?)Subst // Subst =Lipid A AG: |
If known, the linking atom in the aglycon is specified the following way:
AG: (->3) 2,3-dihydroxy fatty acid (a fatty acid with unknown length/saturation degree is attached via hydroxyl at its C3).
In O- and N-glycans, encode aminoacid in ST1 if it is known, or write a general rule in AG othrewise. Please note modifier 1N in N-glycans reflecting that there is no -OH group at the attachment site:
| ST1: aDGlcp1N(1-4)xLAsn AG: protein CC: N-glycan |
or | ST1: aDGlcp(1-3)xLThr AG: protein CC: O-glycan |
or | ST1: aDGlcp AG: (->3) L-Thr/L-Ser (protein) CC: O-glycan |
In all other glycosides, than O-linked, use prefices/siffices at the anomeric position to indicate elimination of the -OH group: 1N suffix for N-glycosides; 1S suffix for S-glycosides; 1d prefix (or 1, in case there are more deoxygenated carbons) for C-glycosides, e.g. bDGlcp1N(1-4)xLAsn; bDGlcp1S(1-3)xLCys; bD1dGlcp(1C-3)Ph; bD1,2,6daraHexp(1C-1)Subst // Subst = ethane = SMILES {1}CC
Atom numbering is used to derive substitution positions. For residues in the CSDB vocabulary it should follow the atomic pattern specified in Monomeric namespace. In simple cases like common monosaccharides or amino acids it corresponds to IUPAC recommendations.
For atypical aglycons and Subst aliases, you can use any numbering scheme, however substitution positions in CSDB Linear (ST1 field), and in SMILES (in curly brackets), and in NMRH/NMRC data prefices should match each other. Usage of atom numbering common for compounds/residues of the same class is recommended. If it is missing or unobvious, use the same numbering as provided by the authors of the annotated paper. It is also advisable to use the same numbering scheme for similar aglycons in different records.
The unit type is stored separately in the ST2 field and can be the following:
Chemical vs. biological repeating units. A chemical repeating unit does not imply knowledge of the repeating frame positioning, e.g. -ABC-, -BCA-, and -CAB- are the same polymers. In most papers where a primary structure of glycopolymers is elucidated, authors do not track the end groups, and thus provide a structure of a chemical repeating unit (CHEM) only. In contrast, if authors determine what exact residues occupy the reducing and non-reducing ends of a polymer (e.g. if they know from the biochemical context by which residue the polymer is attached to the core or other carrier), the three above variants are NOT the same structure anymore, and in this case, the declared repeating unit is called a biological one (BIOL). Sometimes biological repeating unit is not proven experimentally but it can be suggested (SBIOL) basing on the same structure from the same organism in some other record (BIOL) or on general knowledge, e.g. if a repeating unit contains a single aminosugar, its biological repeat is likely to start with this aminosugar.
Polymerization degree should be specified in ST3 field if known. Ranges, lists and relation symbols are supported, e.g. ST3: n=5-7, 12 or ST3: n>100. The number indicates how many types the whole structure is repeated; for subrepeating units inside the structure see section Repeating parts.
The n character tells that the value is a polymerization degree. If there is no n character, the value is interpreted as molecular mass in Daltons, e.g. ST3: 1686.3 [M+H]+ (hydrogenated molecular ion) or ST3: 70000-100000 (70-100 kDa).
Some structures can be recorded in multiple ways, especially when a full record includes other fields than ST1 (CSDB Linear code). If selection rules are not defined, please use the following criteria: