This page will generate a Hamming distance matrix between mono- or dimeric fragment pools from taxa populated in CSDB. The matrix will be normalized by taxon coverage and used for clustering of taxa, visualized as phenetic trees, and exported for external processing.
| Scope settings |
|
Limit taxonomical scope to:
|
| General settings |
|
|
Rank of taxa to compare (should be lower than selected scope).
Specify exact species (all)
|
|
|
Taxon population threshold. Minimal number of structures assigned to a taxon or its subtaxa, to include this taxon in calculation (affects selection of taxa). Check to use this filter. |
|
‰ |
Normalized taxon population threshold. Minimal part of structures assigned to a taxon or its subtaxa, to include this taxon in calculation (affects selection of taxa).
The part is calculated against the total number of structures from organisms of the same kingdom. Check to use this filter. |
|
|
Structure abundance threshold. Minimal number of structures in which a fragment should be contained to be qualified as 'present in biota' (affects selection of fragments) |
|
|
Fragment abundance threshold. Minimal number of instances in which a fragment should be present to be qualified as 'present in biota' (affects selection of fragments) |
|
|
Fragment presence threshold. Minimal number of instances in which a fragment should be present in organisms of a taxon to be qualified as present in this taxon (affects occurence codes and thus, taxon dissimilarity) |
|
|
Size of fragments to analyze (dimeric or monomeric) |
|
|
Type of structures to analyze. Only structures of this type are considered in fragment analysis and where marked by (*). 'Optimized' = only polymers from bacteria, archaea and fungi, and only mono/oligomers from plants. |
|
|
Format of the dissimilarity matrix |
| Fragment pool generation settings |
|
|
Combine anomeric forms. All sugar residues will be treated as 'any anomer' |
|
|
Exclude underdetermined residues. Residues with unknown anomeric, absolute or ringsize configuration will be omitted from analysis. |
|
|
Exclude monovalent residues. Residues like Me, Ac, etc. will be omitted from analysis. Please note, that Ac in N-acetylated aminosugars is a separate residue. |
|
|
Exclude superclasses. Fragments with residues represented by aliases and superclasses will be omitted from analysis. |
|
|
Differentiate aliases. Residue aliases (used for atypical residues) will be differentiated by actual residue names, otherwise they are combined under an alias name. |
|
|
Sugars only. Fragments with non-sugar residues (including monovalent residues, like N-acetyls) will be omitted from analysis. |
|
|
Exclude aglycons. Fragments with atypical residues at non-reducing ends will be omitted from analysis. |
|
|
Differentiate location. The same fragments at different locations (inline, terminal, reducing) will be treated as different. |
|
|
Strict comparizon of fragments. Unknown configurations and ringsizes are always unequal to those known (otherwise a fuzzy comparizon is performed). |
|
Please, enter this code: 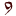  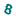 --> -->
(may take up to 10 minutes)
|
