CSDB usage: basic operations
This section describes details of basic user operations in CSDB. For additional help, refer to Extras and Maintenance help.
Content:
Menu
The menu is what you see on the left side of the screen when you enter the project web-site. If you don't see the menu, the frame support is probably turned off in your browser.
Red numbers indicate how many distinct structures are currently present in CSDB and in how many publications. Four sections stand for the following operation groups:
Performing the queries
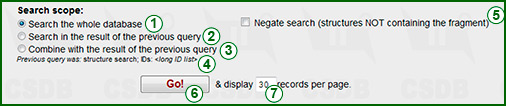
In every search form you are expected to fill in the search terms, select the scope and run the query. The search terms depend on the type of search and are explained in the subsequent sections. The Go button (6) processes the query. Text field (7) informs the search engine how many result records should be output per page (default is 30).
Every search form, except the ID search, has a selector identifying the search scope. This feature allows refining the queries by their intersection or combination with queries of different types. If the default value whole database (1) is selected, the search will be performed through the whole database content.
If there has been a previous query within the current browser session, two more variants become unblocked: Search in the result of the previous query (2) and Combine with the result of the previous query (3). A summary of the previous query results (4) is displayed below the selector.
Search in... intersects the current and previous queries (logical AND), e.g. you can first search for some substructure and then refine the results using some bibliographic data by selection of the search scope as (2).
Combine with... combines the current and previous queries (logical OR), e.g. you can first search for some substructure and then extend the results using some other substructure or other data by selection of the search scope as (3).
If the Negate search checkbox (5) is checked, the current query will be negated (logical NOT), i.e. the database returns all results that do NOT match the search criteria. This option applies to the results after processing all other search-dependent options, so use it carefully if multiple search criteria are specified. Combining this option with Search in... produces the logical AND NOT operation.
Note: if you intersect or combine search results of different type more than twice, at the final step you may obtain more results than expected. For example, imagine that you searched for the substructure "bDQuipN4N" and got 100 structures (compound IDs). Then you searched for publications using AND scope, applied the criterion "published after 2005", and got 20 papers (publication IDs) containing the structures from the previous query. After this you searched for organisms using AND NOT scope, specify "Shigella" genus, and got 1000 taxons (organism IDs), which is much more than you expected. This is because the criterion "AND NOT Shigella" was applied only to the results of the previous search with no regard to pre-previous search. It means that returned results are those organisms, which do not belong to the Shigella genus and which are published in the 20 papers found at the second step. However, those 20 papers might contain other structures (not only those structures containing "bDQuipN4N" which you found at the first step) assigned to other organisms, giving you 1000 taxons in total. This behavior is applicable to search series in which you intersect results three or more times, and at least two steps use a search type different from the previous one (e.g. "structures AND articles AND organisms", or "structures AND articles AND structures"). Series like "structures AND structures AND articles AND articles" are not subject to this behavior because there is only one variantion of a search type.
Search using IDs
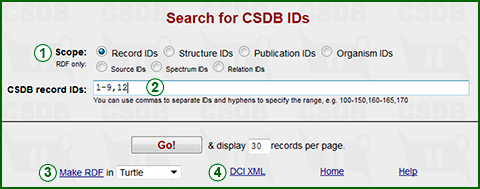
This form allows data retrieval using unique CSDB identifiers for record, publication, compound or organism. The record ID is equal to the value of the ID field in the CSDB dump file. Enter ID or ID range in text field (2), separating different IDs or ID ranges with commas, e.g. 1,2. The range is identified by hyphen, e.g. 1-10.
By changing the search scope (1), you can search for other CSDB IDs: structure IDs, publication IDs and organism IDs. The remaining IDs (source, spectrum, relation) are proposed for RDF generation only (you cannot use them for search queries).
In most cases, users do not know these IDs. The ID search may be useful for fast access to the data that have been previously searched for, if the ID was remembered. To make a link to a certain record from a webpage, you should use the following syntax <A HREF="http://csdb.glycoscience.ru/{DB_name}/core/search_id.php?id_list={my_ID}&mode={my_search_scope}">{my_link_name}</A>, where {my_search_scope} = record (default) or structure or publication or organism, and {DB_name} = bacterial or plant_fungal.
Link (3) produces an RDF feed for selected IDs, link (4) produces meta-data in the ThomsonReuters DCI XML format.
(Sub)structure search
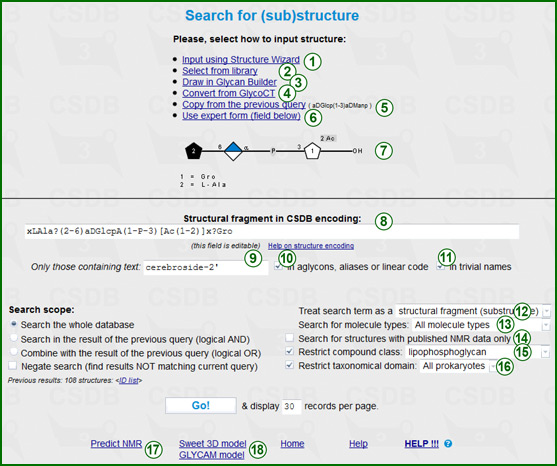
This form lets you search the database by fragments of chemical structure. As you enter the structure by one of these methods, pressing Return the structure to the search page... returns you back to the structure search form with the pre-filled search term field (7). The result of your input is converted to the CSDB linear encoding; it is put to field (8). This field remains editable, so you can use the generated structure as a starting point for manual editing. As soon as CSDB linear encoding is present, the structure is previeved in area (7) in the graphic SNFG format. If the structure was entered manually but could not be parsed, parsing errors are displayed instead.
There are several ways to input the structure term:
- Input using the structure wizard (1). The usage of this variant implies construction of a structure by visual operations and does not require special knowledge except general nomenclature of carbohydrates. On the other hand, it has some limitations, i.e. not all the queries, which can be processed by the search engine, can be constructed by the wizard. For help on the structure wizard, please refer to the next section.
- Select from library (2). This option opens the window to choose a widespread structure by its common name. The selected structure is previewed in a pseudographical format before use.
- Draw in Sugar Sketcher (3). Sugar Sketcheris a web application for graphic input of simple carbohydrate structures in the SNFG notation.
- Convert from GlycoCT (4). This variant accepts the structure in the GlycoCT extended format, converts it to the CSDB linear encoding, reports conversion problems if any and returns the structure in the proper format. The converted structure is previewed before use.
- Copy from the previous structural query (5). This option is available only if there has already been a structural query within your browser session. It copies the previous structural query to the search term field, so you don't have to re-enter the whole structure in case of minor changes.
- Use expert form (6) focuses input to the search term field for manual typing. Usage of this variant implies knowledge of the CSDB structure encoding rules. In the search term, you can mark the residues that should occupy terminal (non-reducing) positions in structures by preceeding ^^. To restrict a residue as terminal but still allow monovalent substituents (Ac, Me, etc.), use ^ prefix. For example, to find structures containing an acetylated or non-acetylated 1-3-glucosaminoglucan disaccharide at terminal position, search for ^?DGlcpN(1-3)?DGlc?
Literal term (9) limits the search to those compounds that have the specified text in their CSDB linear encoding (including aglycons and 'Subst' aliases). Two checkboxes define where to look for the text: in aglycons, structural annotations and aliases and structrure linear codes (10), and/or in trivial names of compounds (11). If no structure is specified, but the literal term is given, all structures matching this text will be returned, as if the structure was ANY.
By default, the query engine interprets your term as a substructure, i.e. those structures will be returned that contain the specified fragment (including fragments located across polymer repeating unit borders). Interpretation of the anomeric, absolute and ringsize configurations of residues depends on the strictness level selected in Treat search term as... selector (12). Implicit (default option) means that configurations are matched in a fuzzy way, e.g. aLGlcp query will find a?Glc? together with strict matches.
Explicit means that structures containing underdetermined residues which CAN match the query, will not be returned, e.g. aLGlcp query will find Glc in explicit a,L,p-configuration only.
However, upon a search for an underdetermied form (??Glc?), both explicit and underdetermied forms will be returned anyway.
To limit search only to those structures that match the search term exactly (are equal, rather than contain) use Treat search term as... selector (12) to choose complete structure. Open linkages in the search term are interpreted differently depending on the status of this option. In case of search for a fragment, open linkages are considered links to ANY residues, e.g. -3)aGlcp(1- is equivalent to the ANY(?-3)aGlcp(1-?)ANY fragment. In case of search for a complete structure, open linkages indicate the repeating unit of a polymer, e.g. -3)aGlcp(1- is equivalent to [-3)aGlcp(1-]n.
In case of search for the complete structure of a heteropolymer, only those records will be found where this polymer is described using the same repeating unit frame as in the search request. If the complete search term is a polymeric structure, two result sets will be combined:
- biological repeating units that have exactly the same repeating unit frame positioning
- chemical repeating units and motifs that have this or another repeating unit frame positioning (i.e. shift of the repeating unit frame does not make a structure different)
The comparison of complete structures may be strict or fuzzy, accordingly to the selected option. The strict comparison implies exact match between the search term and the structure, except the repeating unit positioning in polymers.
The fuzzy comparison allows the following deviations between the search term and the matching results:
- Structures with stoichiometry of residues different from that in the search term will be found (e.g. Glc-Gal --> %Glc-Gal, and vice versa)
- The structures with unknown anomeric, absolute or ringsize configurations will be found, if they do not contradict the search term (e.g. aDGlcp --> aDGlcp, ?DGlcp, aDGlc, a?Glc)
- If a question mark is specified for the anomeric, absolute or ringsize configuration, structures with both unknown and any known configurations will be found (e.g. ?DGlcp --> ?DGlcp, aDGlcp, bDGlcp)
- The structures with unknown linkage positions will be found, if they do not contradict the search term (e.g. Glc(1-2)Gal --> Glc(1-2)Gal, Glc(1-?)Gal, Glc(?-?)Gal)
- If a question mark is specified for the linkage position, structures with both unknown and any known linkage positions will be found (e.g. Glc(1-?)Gal --> Glc(1-2)Gal, Glc(1-?)Gal)
- Structures with alternative parts will be found if at least one possible combination of alternatives matches the search term (e.g. Glc-Man --> Glc-Man, <<Glc|Gal>>Man, <<Glc|Gal>><<Rha|Man>>)
- If a search term contains alternative parts (angle brackets), it is split into all possible combinations of alternatives regarding the angle bracket logic (inclusive or exclusive), each of the resulting structures is used for the search, and results are combined (e.g. Rha<<Glc|Gal>>Man --> Rha-Glc-Man, Rha-Gal-Man; <Ac(1-2)|Ac(1-3)>Man --> Man2Ac, Man3Ac, Man2Ac3Ac)
- Structures with internal repeating parts will be found if the number of repeats can match the search term (e.g. n=4 --> n=4, n=?, n=3-5; n=3-5 -> n=3, n=4-10). In complete structure mode Structures where repeating parts are recorded as multiple entities (Glc-Glc-Glc instead of /Glc/n=3/ ) will not be found, and vice versa.
The Search for molecule types selector (13) specifies among which structure types the search is performed (monomers, oligomers, polymers, biological repeating units, fragments/motifsm etc.). All molecule types (default) means no limitations.
The Search for structures with published NMR data only checkbox (14) limits the search only to those compounds that have an NMR assignment table stored. The Compound class checkbox and drop-down list (15) limit the search to a certain compound class. The Restrict taxonomical domain checkbox and drop-down list (16) limit the search to structures assigned to organisms from the certain taxonomic kingdom.
There are additional tools, which do not process the query:
The Predict NMR link (17) simulates a 13C and 1H NMR spectra for a given structure. For more information, please refer to the NMR prediction help.
The Sweet 3D model link (18) sends te structure to Sweet-II engine for 3D visualization. The Glycam model link (18) sends the structure to AMBER/GLYCAM for conformational processing and generation of atomic coordinates.
Structure wizard
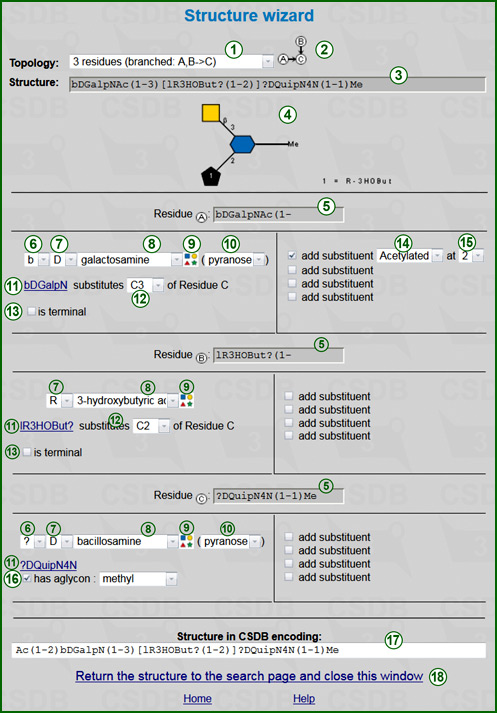
The wizard allows construction of structural queries without knowledge of the CSDB structure encoding notation. The first step is to select a topology from drop-down list (1). The wizard supports topologies of up to four residues (besides monovalent substituents). The graphic representation of topology (2) is displayed on the right. As soon as topology is selected, the appropriate number of residue sections appears below (three on the screenshot). The one-line preview of the structure (3) and its SNFG image (4) are displayed below the topology.
Each residue section header (5) includes a position of this residue within the selected topology (e.g. Residue A) and the constructed residue name with all configurations and substituents applied.
Each residue section includes the following controls:
- The anomeric configuration selector (6) is displayed if currently selected residue can have different anomeric configurations. Visibility if this field depends on the selected residue name. Possible values are a(alpha), b(beta) and ?(any).
- The absolute configuration selector (7) is displayed if currently selected residue is chiral. Visibility if this field depends on the selected residue name. Possible values are D or L for multichiral residues, R or S for residues with only one stereocenter, and ? (any).
- The residue name drop-down list (8) selects the residue name. Only the widespread residues, which are present in more than 30 structures in CSDB, are listed. If a residue of interest is missing from this list, please select its first item (complete list). This operation pops up a complete list of residues supported by the database. A simple form in this window allows construction of a residue from its name, anomeric, absolute, and ringsize configurations (if applicable); on close it copies the result to the structure wizard.
Alternatively, you can select the residue in graphic form from the SNFG icon map displayed by clicking on icon (9).
The blue link (11) is the resulting residue name with configurations applied but without substituents. This link leads to the complete list and is updated as soon as you change any parameters of the residue.
- The Residue type drop-down list (10) selects the residue type (pyranose, furanose, open-chain, alditol, or ? for any). It is visible only for monosacharide residues.
- The linkage control (12) informs the wizard which position of the other residue is substituted by the current residue. E.g. substitutes C3 of residue C means that this residue (donor) bonds to C3 of residue C (acceptor). The term residue C in this example is automatically set according to the selected topology, while C3 is selected by user. Variants from C1 to C9 and ? (any position) are available. The position of the current residue by which it substitutes the other resdiue is calculated as 1 (for aldoses and other residues) or 2 (for ketoses or N-linked amino acids).
The residue at the reducing end of the fragment does not have this control.
- The is terminal checkbox (13) indicates that this residue is at non-reducing terminus, i.e. it cannot be substituted by any substituents except monovalent ones. This checkbox is visible only for the residues occupying terminal positions in the selected topology.
The residue at the reducing end (the last residue section) has the aglycon selector (16). If checked, an aglycon can be selected from a drop-down list; otherwise there are no aglycon restrictions. Please do not check this checkbox for polymer repeating unit structures.
- Each residue section has four Add substituent controls on the right. They allow specification of up to four monovalent substituents. When checked, drop-down lists (14) and (15) are displayed and identify monovalent substituents and positions of their attachment, accordingly. E.g. on the screenshot residue A (GalN) is acetylated at position 2 to form GalNAc.
The resulting structural query term (17) is displayed at the page bottom. By pressing Return the above structure to the structure search page... (18) it is passed to the parent window (usually a structural or compositional query form), and the wizard is closed.
CSDB/SNFG structure editor
The CSDB/SNFG editor is a graphical tool allowing construction of an arbitrary glycan or a glycopolymer by intuitive operations with SNFG icons. The output is a search term in CSDB Linear notation and exportable images. Version 1β provides the following features:
- all non-cyclic oligomeric and polymeric topologies;
- over 500 residues supported by CSDB, including superclasses and non-carbohydrate constituents;
- aliases for unsupported residues with SMILES specification of structure;
- name- or abbreviation-based search for residues and their modifications; quick access to popular constituents via toolbars;
- specification of bond donor and acceptor; regular, chelate and carbon-carbon bonds;
- anomeric, absolute, and ring size configurations of monosaccharides; absolute configuration of alditols and non-carbohydrate constituents;
- uncertainties at the level of residue identity, substitution pattern, residue configurations;
- uncertainties at the topological level: sets of alternative branches combined exclusively (XOR) or inclusively (OR);
- repeating substructures including nested repeats;
- instant preview in several text notations and in SNFG;
- instant preview of chemical structure in SMILES notation, and as a structural formula;
- preview of the optimized 3D molecular structure with multiple rendering and export options;
- export to CSDB Linear, WURCS, Glycam, SweetDB, GlycoCT, SMILES, MOL, and PDB formats, and to hi-res SNFG images (SVG or GIF);
- import from CSDB Linear and GlycoCT notations;
- availability as a module for external usage in your project.
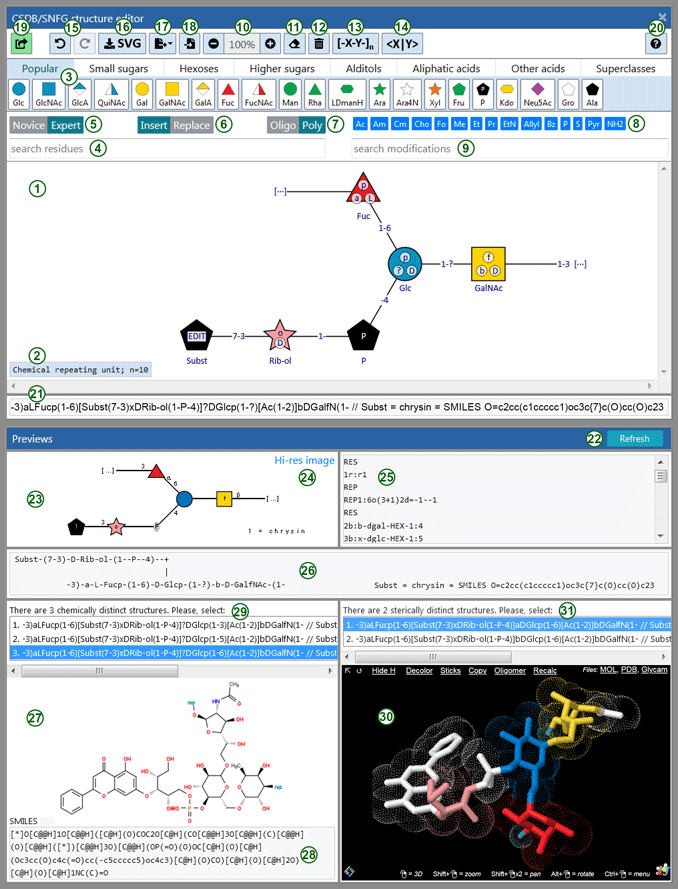
The user interface includes a drawing canvas (1), toolbars (3-20), CSDB Linear preview (21) and other previews (22-31).
Drawing canvas is an area (1) where your structure is sketched. To start structure input, click on any icon in the toolbar to add a residue to the sketch. The first added residue becomes a reducing end of an oligomeric glycan or a glycoconjugate. Subsequently added residues are appended as donors to a residue currently selected. The selection is indicated by red highlight around a residue. Only one residue in the sketch can be selected; to select another one click on it. The last-appended residue becomes selected automatically. To create a branched topology several residues should be appended to the same acceptor (branching point). To do so, select the branching point again after adding each new branch.
For your convenience, some residues (like Ac, Me, etc.) can be alternatively added as monovalent modifications of other residues. To avoid cluttering, such modifications are displayed below residue icons in the sketch, together with their attachment positions.
Any residue in a sketch can contain up to three controls, displayed as semi-transparent disks in front of an SNFG icon: absolute configuration, anomeric configuration and ring size. Clicking these disks sequentially loops through all parameter values applicable to the residue. If there is only one value possible, the corresponding control is hidden. For example, if a residue is achiral or its chirality is encoded in its name (e.g. Kdo or LDmanHep) the absolute configuration disk is not displayed. Similarly, anomeric configuration and ring size disks are displayed only for monosaccharides that can have a cyclic form.
Residue caption in a sketch includes residue abbreviation (first line) and a list of modifications and their attachment positions (other lines, if present). Clicking on a residue caption opens a modification dialog (see below).
Residues on a sketch are connected by bonds and each bond has a caption displaying the linkage positions in the donor and acceptor residues. By default, the newly-added residues are connected via default outgoing position (1 for aldoses, 2 for ketoses, etc.) and unknown (?) ingoing position. Clicking on a bond caption opens a bond dialog (see below), where you can change linkage positions and other bond options.
Upon change of some parameters (residue identity, deoxygenation pattern, ring size, linkage and modification positions) their combination can become chemically impossible. In such cases other parameters are automatically adjusted to retain the validity of the structure, and a warning message is displayed. Atom types and numbering are tracked to ensure that all inter-residue bonds, except specially marked carbon-carbon ones, are formed with elimination of water (typically, hydroxy → hydroxy, hydroxy → carboxy, amino → carboxy).
You can zoom the canvas by zoom control (10). The value between - and + buttons indicates the current zoom level. If a structure is to big to fit in canvas, scroll bars are unblocked. You can pan the sketch by click-and-drag anywhere on the canvas but not on the structure.
Structure type and additional attributes (polymerization degree, molecular weight) are displayed in the lower left corner of the canvas (2), if specified. Clicking this panel displays a dialog to change the attributes or refine the structure type basing on the topology in the sketch.
Toolbars include four sections (top to bottom):
- control buttons (10-20);
- eight tabs with most abundant monomeric residues (3);
- mode selectors (operating mode, input mode, structure type) and popular modification buttons (5-8);
- searchable dropdowns for complete lists of residues and modifications (4,9) (available in the Expert mode).
The editor has two operational modes, selectable by control (5) (Novice/Expert, selected state has aqua color). In the Expert mode all editing features are enabled. The Novice mode simplifies the interface but has the following limitations:
- only the most abundant residues (3) and modifications (8) can be drawn, and complete lists of residues and modifications (4,9) are hidden;
- absolute configurations, ringsizes and outgoing bond positions are assumed to be the default ones for each residue and cannot be edited;
- rarely occuring bonds (amino/amide → amino/amide, amino/amide → hydroxy, hydroxy → amino/amide) cannot be drawn.
Monomeric residues most abundant in natural compounds are presented in toolbar (3) and divided into several categorical tabs: popular residues of all types, abundant hexoses, small sugars, higher sugars, fatty acids, amino- and other acids, residue superclasses. Every button displays an SNFG icon and an abbreviation, while more detailed descriptions are available on the tooltips shown when you hold a mouse over a button. Some buttons present popular combinations of residues, such as GlcNAc (glucosamine + acetic acid).
Popular modifications (small residues that are no further substituted) are presented in a toolbar (8). Clicking on a modification button opens a modification dialog (see below).
If a residue of interest is missing from toolbars, start typing its name or abbreviation in field (4) (Search residues) to locate it in the complete list of residues. As you type two or more symbols, matching entries are shown in a dropdown list, where you can pick an entry to add it to the sketch. Analogous feature for modifications is available via field (9) (Search modifications).
Depending on control (6) (Insert/Replace, selected state has aqua color), you can change how the new residues affect the sketch. In the Insert mode (default) residues are added as new donor branches to the currently selected residue. In the Replace mode a new residue replaces the currently selected one.
The sketched structure can be interpreted as an oligomer or as a regular polymer repeating unit. The latter case implies one more bond between the repeating units, which is displayed as two open bonds at the left and right termini of the structure. The [...] mark indicates the next/previous repeating unit instance. To switch between polymers and oligomers, use switch (7) (Oligo/Poly, selected state has aqua color). Activation of a Poly mode sets the selected residue as an acceptor (left) terminus of the repeating unit. Reducing end (root) of the current structure becomes a donor (right) terminus automatically. Activation of an Oligo mode removes the hanging bonds at both termini and converts the structure to an oligomer.
Clear button (11) deletes the entire structure and you can start anew. The same effect is when you delete a root residue. Delete branch button (12) deletes a part of the structure including the selected residue and all of its branches (ingoing substituents, donors).
Subrepeat button (13) opens a dialog to mark a part of the structure as an iternal repeating unit (see below).
Alternative button (14) appends a block of alternative branches to the sketch, as a special residue named Fuzzy. Use EDIT control displayed over a Fuzzy residue icon to open a dialog to define alternative branches (see below).
Undo/Redo buttons (15) cancel the latest operation or restore the canceled action correspondingly. Undo button becomes disabled, when you reach a maximal undo depth (10 steps by default).
Export button (17) opens a menu to select the export format.
Supported carbohydrate notations are: GlycoCT, WURCS, GLYCAM, SweetDB. On one of these formats selected, the exported structure is displayed in a small window where you can copy it to clipboard. These formats support not all of the features that you can draw in the editor, so some limitations exist (see error messages for details).
Supported general chemical formats are: SMILES, MOL, PDB (with residue naming). On one of these formats selected, an export dialog is displayed to download a corresponding text file. Atom coordinates are optimized before export in MOL or PDB formats. For underdetermined structures that can have multiple structural or stereo isomers selectors are displayed to pick a certain variant. The first variant is All structures; it exports multiple data blocks in one file.
Import button (18) opens a dialog where you can type or paste a structure for import. You have to chooze the format of the import term: CSDB Linear(default) or condensed GlycoCT. The existing structure is replaced by the imported one.
When your structure is ready and correctly previewed, click Done button (19) to close the editor and pass the structure to the parent search form. The CSDB Linear code (21) prepared from your sketch appears in the editable field of Substructure search as a search term.
Help button (20) toggles small question marks beside every major control. Further you can click on a corresponding question mark to get a quick help on the matching element.
Preview areas below the canvas show how the sketch is processed to generate the structure.
The CSDB Linear preview (15) displays the current structure in CSDB Linear notation and is updated upon any change of a drawing.
Other previews section (22) displays the current structure in various carbohydrate and chemical notations. These previews are updated silently in several seconds after each change of the structure. The countdown to the next update is displayed in the header of the section. To skip time-out and update previews immediately, click Refresh button on the Previews header panel. In case of error (unparseable structure, chemically impossible bonds, incompatible configurations, etc.), an error message is displayed instead of previews.
The graphical preview (23) displays an SNFG image returned by CSDB server after processing the generated CSDB Linear code. The graphical preview is updated immediately upon any change of a drawing and is mainly intended to make sure that the drawn structure could be correctly recognized.
Hi-res image link (24) opens a page to export the generated SNFG image as a high-resolution raster file to be pasted in your publication. You can select a background, including transparent, a scale factor, and a few other display options. The sketch itself can also be exported as a vector (SVG) image by clicking on Export SVG button (16) in the upper toolbar.
GlycoCT preview (25) displays a GlycoCT condensed code obtained by conversion from the generated CSDB Linear term.
SweetDB preview (26) displays a pseudographic representation (SweetDB format) obtained by conversion from the generated CSDB Linear term.
Formula and SMILES preview displays a structural formula (27) and a SMILES code (28) of the drawn structure. If the structure contains chemical uncertainties, all possible chemically distinct isomeric CSDB Linear entries will be listed in selector (29). The topmost item is selected by default.
3D preview (30) displays a 3D molecular view of a MOL-file generated from CSDB Linear entry currently selected in Formula and SMILES preview: once the isomer is selected (or changed), Show 3D button appears in the 3D preview area. To avoid lags, the data are loaded into the applet only when this button is pressed. If selected isomer contains sterical uncertainties, all possible stereo-isomers are listed in a selector (31). The topmost item is selected by default. Top line of the 3D viewer has controls for display options and links to download a displayed 3D structure as a file in the following formats: MOL, PDB with classic residue naming, and PDB with GLYCAM residue naming. To access full functionality of 3D structure rendering, please use a Jsmol menu (right-click on the applet).
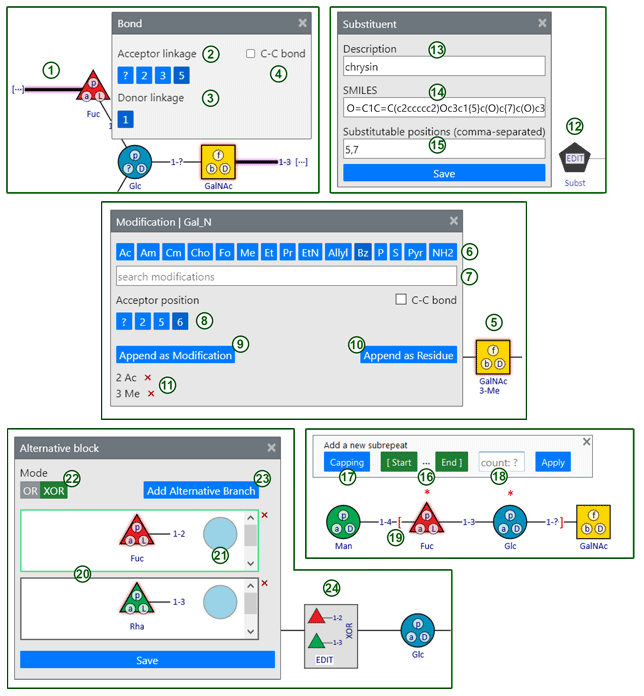
The modal dialogs allow modification of structural features, and prevent other user actions until they are closed. To close a dialog, use a cross in its upper right corner. The numbered elements below refer to the second figure.
Bond dialog is opened when you click on a bond caption. The currently selected bond (1) is highlighted with red glow. Depending on the linked residues, corresponding lists of available linkage positions are displayed. Acceptor linkage (2) is a bonded position in the acceptor residue (usually on the right). Only vacant positions (not occupied by other bonds and modifications) are available for selection. Donor linkage (3) is a bonded position in the donor residue (usually on the left); for monosaccharide residue it is an automatically selected anomeric atom.
Additional controls (4) include:
- a carbon-carbon bond checkbox (for C-glycosides or atypical monosaccharides with branched carbon skeleton);
- a dual bond checkbox displayed if selected donor position can form a chelate linkage with two positions in the acceptor (e.g. carbonyl atom prone to diacetal formation). With this checkbox checked, two donor positions can be selected by clicking them sequentially.
Modification dialog is opened when you add a modification from a toolbar or a searchable list, or when you click on a caption below a residue of interest (5).
To add a modification, click a button with a corresponding abbreviation on a button bar (6) or search for modification name/abbreviation in the complete list (7), then select the attachment position (8) in the acceptor residue. Only unoccupied attachment positions are listed. Clicking Append as modification button (9) adds the modification to the residue. Clicking Append as residue button (10) adds a selected modification entity as a new separately drawn residue.
All modifications of the residue are listed at the bottom of the dialog (11). To delete a modification, click a red cross on the right of the modification name.
Alternatively, a selected residue can be modified by clicking on a blue button in a group of popular modifications in the upper toolbar or by selection from a searchable dropdown list. In this scenario the modification dialog is opened with preselected modification, and you have to select a modification attachment position only. If a modification is linked to a carrier by more than one bond, it should be added as another residue.
Substituent dialog is proposed for adjustment of an atypical residue missing from CSDB vocabulary. Such residues can be added using a Subst button on a Superclasses tab.
This Subst alias for atypical structural entities has specific properties. To specify them, use EDIT control displayed in front of Subst residue icon in the sketch (12).
When using Subst, you have to specify its name in the description field (13), provide a SMILES code with substituted atoms preceded by their numbering in curly brackets (SMILES field (14)), and comma-separated list of substitutable positions in field (15). In some special cases SMILES code can be omitted. More details on the usage of SMILES in CSDB Linear notation are available in the dedicated section of the Structure encoding rules.
Subrepeat dialog is opened when you click subrepeat button on the top toolbar. To define borders of a repeating fragment of structure, select a residue on the sketch and click a corresponding (start for the left end, end for the right end) button (16). Once a terminus is defined, residues that can be selected as another terminus are marked with a red square bracket. To change an external linkage at the left of the subrepeat (start), click capping (17) to loop through the allowed donors (residues or incoming bonds). To add another branch inside a subrepeat, apply changes, exit a subrepeat dialog, select a carrier residue on the sketch, and add a branch regularly. Number of repeating units in a subrepeat should be entered in the input field (18) (default value ? is assumed if this field is empty). To save a subrepeat and append it to the sketch click Apply button. Once a subrepeat is added, you can only edit the number of repetitions. To change a topology of a subrepeat, delete it (click on the number of repeating units and press Delete button) and create a new one.
This feature is purposed for insertion of repeating parts into larger structures. To make the whole structure a repeating unit of a polymer, please use Oligo/Poly switch.
Alternative block dialog is opened when you click EDIT control displayed in a block of alternative entities (24) in the sketch. The dialog allows addition/removal of the alternative branches (20), and their editing in the same manner as in the main sketch. The active branch subject to any commands from upper toolbars is enframed in green (20, upper). To activate another branch for editing, click on it. The acceptor residue carrying the block of alternatives is displayed as a rightmost residue (21) in each alternative branch as a semi-transparent non-editable icon for reference only.
OR/XOR switch (22) toggles the logic of combinaton of alternatives (inclusive vs. exclusive OR). To add an alternative branch, click button (23). To remove a branch click a red cross on the right of the branch. To apply changes and update the main sketch, click Save button.
If a block of alternatives contains chains longer than one residue, and the block itself is substituted in the overall structure, special considerations should be taken about the substituent attachment site. More details on the interpretation of alternative substructures in CSDB Linear are available in the dedicated section of the Structure encoding rules.
Composition search
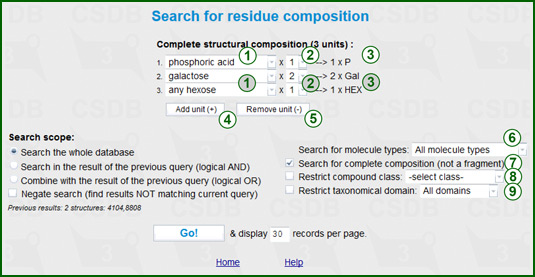
This form allows searching structures by their residue composition, e.g. MS data. The default composition is a single hexose residue. Drop-down list (1) lets you select a residue base type (e.g. HEX, Glc, GlcN etc.) without configurations and ring size. Only most widespread residues are included. If you need a residue, which is missing from this list, you should select its last line - complete list. Control (2) specifies how many instances of this residue should be in the desired composition. The selected residue and its amount are displayed on the right (3).
The first part of the residue list contains superclasses (PEN, HEX etc.). These entries should be used when the unit identity is unclear. Please note, that the HEXN superclass is contained within the HEX superclass, which may produce redundant results when you select HEXN.
Two buttons let you increase (Add unit, (4)) or decrease (Remove unit, (5)) the number of different units in the composition. The total amount of units is displayed in the header. If the composition contains multiple instances of the same type, e.g. two Glucose residues, you should select the residue Glc using (1) and its amount 2 using (2), rather than select 1 x Glc twice.
The Search for molecule types selector (6) specifies among which structure types the search is performed (monomers, oligomers, polymers, biological repeating units, fragments/motifsm etc.). All molecule types (default) means no limitations.
The Search for complete composition checkbox (7) controls the exclusiveness of the search. If it is checked (default), only the structures that contain no other residues except those specified in the composition will be returned, i.e. the input is interpreted as the composition of a complete molecule. If this box is uncheked, the input is interpreted as the composition of a structural fragment, and more structures are returned, including those containing other residues besides those specified.
The Compound class checkbox and drop-down list (8) limit the search to a certain compound class. The Restrict taxonomical domain checkbox and drop-down list (9) limit the search to structures assigned to organisms from the certain taxonomic kingdom.
Structural motif search
The search for structural motifs is based on ranking structures according to how many disaccharides (residues and their linkage are considered using fuzzy logic and without modifications and monovalent substituents) they have in common with the structure in the search term. This feature is not yet supported.
Taxonomic search
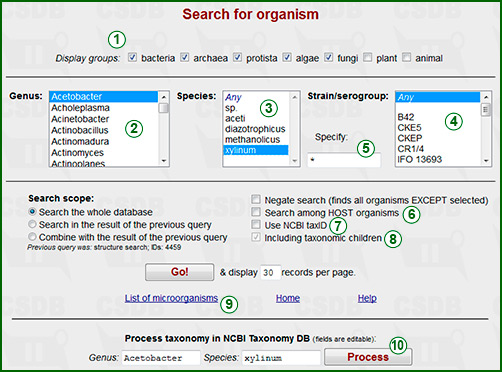
This form allows retrieving particular organisms and associated data by their taxonomic names.
Alphabetical lists of genera (2), species (3) and strains/serogroups (4) provide taxonomic specification (position of an organism in the tree of life). For fast navigation in the genera list, type first characters of a genus name in quick succession.
The lists are generated according to the biological domains selected by the checkboxes in the upper row (1). By default, mainly microorganisms are included (no plants and animals).
The genus (2) should be selected obligatory. Default positions for species (3) and strains (3) are Any which means no limitations. The second position sp. in the list of species is used to find microorganisms with the specified genus and strain but the unassigned species name. As soon as the genus is selected, the species and strain lists are updated accordingly. As the subdivision (strain/serogroup) list may be long, there is text field (5) to enter it directly (or a part of it, to search a taxon by incomplete strain speicfication). The default value is * (=no limitations). If you use a star character, subdivisions are matched as raw strings, e.g. O6* will find O6, O61, H4:O6, etc., otherwise only the tokens with the same numerical index are found, e.g. O6 will find O6, O6a, H4:O6, etc., but not O61.
If the organism was reclassified or a taxon was renamed, you can use any of the names to identify the organism. However, not all of the organism name remappings and synonyms are currently stored in CSDB.
The Search among host organisms checkbox (6) will search the specified taxon among host organisms, rather than among associated organisms themselves. E.g., if you specify Mus musculus and check this box, the search will return this organism and structures that were found in all microorganisms infecting mice or extracted from mice.
On checking Use NCBI Tax ID (7), taxon selection lists disappear, and you can identify an organism or a taxonomic group by the NCBI Taxonomy Database ID. If Including taxonomic children (8) is checked, all organisms that belong to the specified group and have lower ranks will also be included (e.g. search for Proteus with this checkbox on will return Proteus penneri, Proteus mirabilis, Proteus sp. 1234 etc.). If you use selection lists rather than NCBI Tax ID, taxonomic children are always included.
The link List of organisms (9) displays the complete list of organisms that are present in the database. The form Process taxonomy in NCBI... (10) retrieves the data from the NCBI Taxonomy Database using the selected genus and species as criteria. The data displayed are: scientific name of the organism, synonymic names, rank and taxonomic lineage.
Bibliographic search
This form is proposed for search using bibliographic data and keywords. If search criteria are provided in several sections of this form (e.g. authors and title), the intersection of queries will be returned. The queries are case-insensitive and accent-independent (but please note, that accented consonants may be stored as a combination of latin characters in publication metadata).
The Authors field (1) lets you input the author name(s). Click on a character in the helper (2) to input national symbols missing from your keyboard. To avoid spelling errors, it is recommended to use the author index available from the Author index button (3). For the author index window to appear, at least two first characters of the author family name (4) should be specified beside the button. The list is restricted to the author names beginning with the specified character combination. As soon as you click a certain author in this list, it is copied to the bibliographic form. The author field supports query language with term grouping and wildcards (click here for details). The sample query "Armstrong L" AND (Einstein) will find publications written together by Armstrong L and Einstein (with any initials).
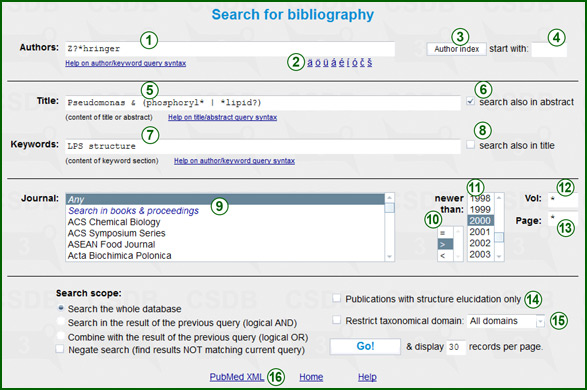
The Title field (5) lets you define which words should be present in the publication title. This field supports query language with term grouping, wildcards and logical operations (click here for details). The sample query capsul* OR C?S will find publications containing at least one of the following words in the title: capsule, capsules, capsular, CPS, COS.
If the checkbox search also in abstract (6) is checked, publication abstracts will be analyzed for the specified terms together with the titles. Please note that not all of the publications within CSDB have abstracts stored.
To limit the search to publications with certain keywords assigned, use the Keywords field (7). The list of keywords assigned to every publication matches the keyword list published in the paper. The query language is supported.
If the checkbox search also in title (8) is checked, the publication title will be analyzed for the specified terms together with the keyword list.
The lower half of the form allows searching by issue data:
- The Journal can be selected from alphabetical list (9). The first position in this list is Any, which means all available journals will participate in the search. Another special position is Search in books & proceedings, which sets the search scope to all books and symposium abstract collections stored in CSDB. If the list lacks the journal you need, this means there are no matching publications in CSDB. For fast navigation among journals, type first characters of a journal name in quick succession.
- List (11) specifies the year of publication. The default is Any (no restrictions). Selector (10) determines how to interpret the year value: you can search for publications of the exact year specified (=), or newer than the year specified (>), or older (<).
- Text-field (12) stands for the volume number. If you don't know it, use the default value * (=any volume). If you are searching for a particular subvolume, specify it in parentheses, e.g. 71(2) will find (volume 71, subvolume 2) but will not find volume 71 if there are no subvolumes in this journal. On the other hand, 71 will find both (volume 71 with no subvolume specified) and (volume 71, subvolume 2).
- The last issue field (13) is the page number filter. Please specify a page number or leave the default asterisk for no restrictions. If specified, only the records that contain the specified page number between the starting and ending pages of publication, inclusively, will be returned.
Checkbox (14) filters out publications with no structure elucidation described. If this option is checked, publications with additional studies on the structures elucidated elsewhere will not be returned.
The Restrict taxonomical domain checkbox and drop-down list (15) limit the search to publications describing organisms from the certain taxonomic kingdom.
Pressing the PubMed XML link (16) converts the specified bibliographic data to the NCBI PubMed XML format and outputs it in a separate window.
Query language
The query language is supported in the Authors, Title and Keywords fields of the bibliographic search form and utilizes the following syntax:
Quotation (") makes terms unseparable, e.g. "structure elucidation" will return data where these words come together. Always enquote a term if it contains blankspaces, i.e. consists of several words that should come together in the specified order, e.g. "Einstein A". Wildcards (* and ?) are not supported inside quotes.
Asterisk (*) replaces any number (including zero) of alphanumeric characters, e.g. Capsule* will return data containing words Capsule, Capsules, Capsule-like etc.
Question mark (?) replaces exactly one alphanumeric character, e.g. ?-antigen will return data containing words O-antigen, K-antigen etc.
The Title field used to search for terms in publication title and abstract supports additional query syntax:
Ampersand (&) intersects the terms using logical AND, e.g. (Term1 & Term2) will return data containing both Term1 and Term2. This is default option.
Vertical bar (|) combines the terms using logical OR, e.g. (Term1 | Term2) will return data containing either Term1 or Term2, or both.
Power sign (^) applies logical NOT to the term, e.g. ^Term1 will return data NOT containing Term1.
Parentheses are used to group subqueries into more complex expressions and to declare preference of logical operations, e.g. (Term1 | Term2) & (^Term3).
NMR data search
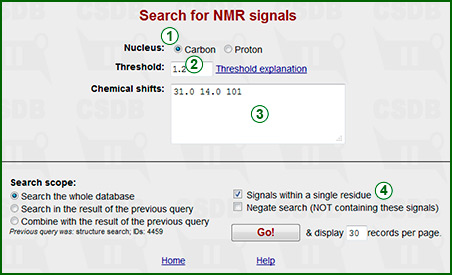
This form allows searching for compounds with NMR spectra containing the specified signals. Selector (1) allows selection of a particular nucleus. The subspectrum to search for should be typed in window (3). You can separate signals with spaces or new line characters, the sorting is not required; the allowed characters are numerals and decimal dot. The threshold field (2) allows output filtering according to the spectra similarity (see below). Only the compounds with the similarity higher than this threshold will be returned. Good values for carbon spectra are 1 and above; good values for proton spectra are 5 and above.
If checkbox (4) is checked (as by default), only the spectra that contain the specified signals within one residue will be returned; otherwise, the spectra are analyzed without assignment of subspectra to residues. Please note, that with this option unchecked the search may take extremely long. Generally, the less signals you specify and the less widespread chemical shifts are, the faster the search is.
To compare the spectra, the CSDB engine forms all possible subspectra of the stored NMR spectrum, the size of the subspectrum being equal to the number of signals in the user input (3). The best-fitting subspectrum is used to calculate the similarity value. Similarity is the estimation of the inverse average deviation between signals, e.g. 1 means the average difference is 1 ppm, 10 means 0.1 ppm, 0.1 means 10 ppm etc. 0 stands for no similarity at all, 1000 is for full similarity (exact match of chemical shifts). If a compound has more than one spectrum (e.g. in different conditions or in different publications), the similarity for this compound is calculated as the average of the similarity values of all spectra assigned to it for given nuclei.
If the user input has less signals than an ideal experimental subspectrum, which may occur due to signal overlap, it will result in lower but still not null similarity. To improve accuracy, it is recommended to input chemical shifts of signals with double integral intensity twice.
The output compounds are sorted in the similarity descending order, and all associated spectra for a given nuclei are displayed. Chemical shifts close to those from the search term (3) are highlighted. The highlight threshold is ±0.4 ppm for carbon chemical shifts and ±0.2 ppm for proton ones.
Output of results
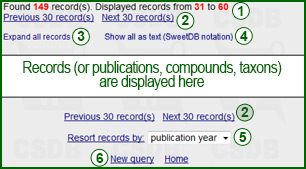
Every search request leads to a number of data units (compounds, publications, organisms etc. ), and what it looks like depends on the type of your search. However, the header and footer (shown on the right) remain similar. Every data unit can be displayed in two forms: collapsed (main data only) and expanded (all related data). Clicking on Collapse this record or Expand this record in the bottom of the data unit switches its display style. Red numbers in the first line of the header (1) represent how many units (compounds, publications, organisms, IDs) have been found and how many of them are displayed on this page. If there are more results than the per-page parameter, the Previous and Next links (2) allow navigation through the result output pages. Expand all records (3) (or collapse all records, depending on the current state) allows bulk operation over the data unit display style.
The footer of the output page contains a link to resort records (4), according to the selected criterion (publication year, microorganism name, etc. - depends on the search type). The resorting is applied to all the records returned, not only to those displayed on the current page. The New query link (5) returns you to the search form of the same type as the one that produced this output.
When you search for a structural fragment or composition or NMR spectra, data units are COMPOUNDS found in CSDB, plus major compound-related data (compound ID, structural formula in the Sweet-DB format, structure type, aglycon, molecular weight, chemical formula, trivial name, NMR data, compound class and references to other structural databases) and a list of publications in which this compound is described. Every publication in this sublist is accompanied with a link to a CSDB record ID (and a list of associated organisms within this record). This record ID covers this compound within this publication and allows access to complete data originating from the paper.
When you search for bibliographic data, data units are PUBLICATIONS found in CSDB, plus major publication-related data (article ID, authors, title, issue data, keywords, publisher, involved institutions, corresponding author's email, used methods and references to other bibliographic databases) and the list of compounds that are described in this publication. Every compound in this sublist is accompanied with a link to a CSDB record ID (and a list of associated organisms within this record). This record ID covers this compound within this publication and allows access to complete data originating from the paper.
When you search for taxonomic data, data units are ORGANISMS found in CSDB, plus major organism-related data (organism ID, systematic name, taxonomic domain, phylum and references to other taxonomic databases) and a list of compounds that are associated with this organism. Every compound in this sublist is accompanied with a link to a CSDB record ID (and an associated publication within this record). This record ID covers this compound in association with this organism and allows access to complete data originating from the paper.
When you search for CSDB record ID or follow the CSDB record ID links in the three previous cases, data units are RECORDS displaying all data available. A record is a combination of compound and a bibligraphic reference, it may have one or more taxonomic references.
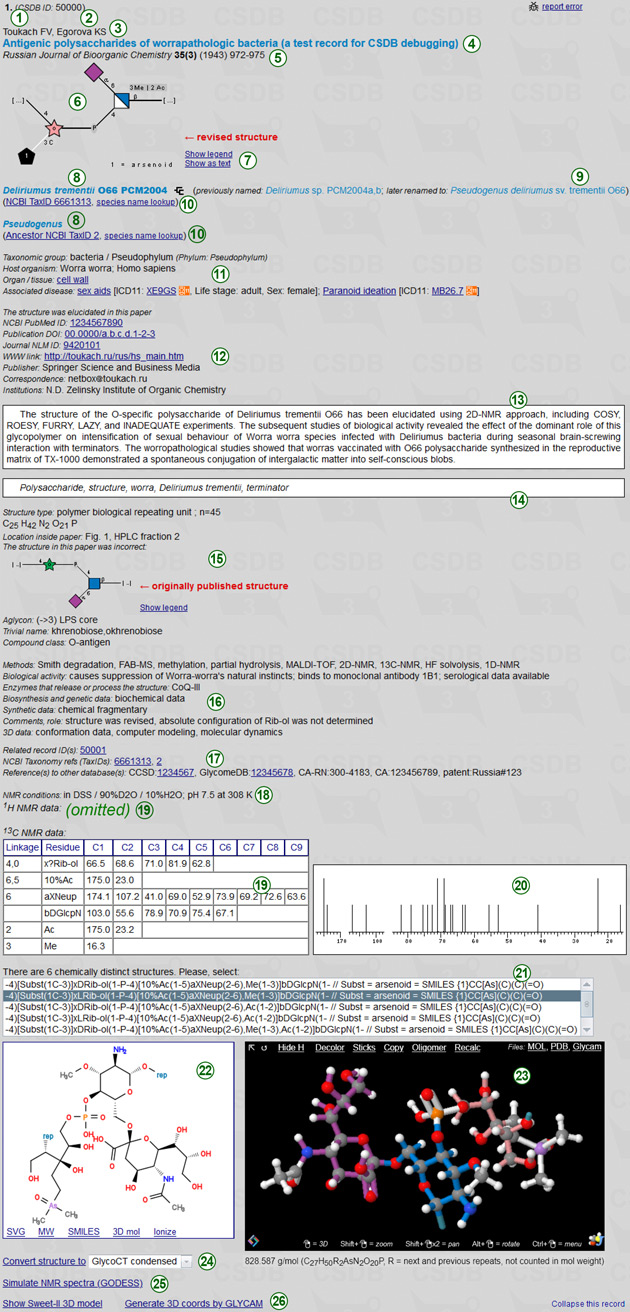
A screenshot on the left shows an example of a completely filled record in the expanded form:
(1) is a number of this record among the records found,
(2) is a CSDB ID of this record.
authors' list (3), article, thesis or chapter title (4) and issue data (5) stand for the bibliographic reference associated with this record. Journal issue data are: journal name, volume, subvolume if any, year, page range. Book and chapter issue data are: book title (incl. series title and volume), editors, publisher, year, chapter number, page range. Symposium proceedings issue data are: symposium name, publisher if available, place, year.
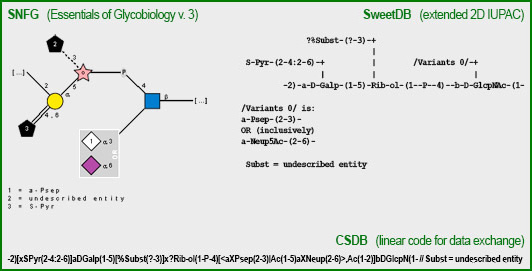 (6) is a graphic representation of the structure in the SNFG format. Clicking on Show as text (7) explains the meaning of the used icons. Clicking on Show as text (7) displays the structure display in pseudo-graphic extended SweetDB format. You can visualy compare the graphic vs. pseudo-graphic representations of the sample structure in the figure on the right. CSDB linear code is not used for visualization; it is given for reference.
Taxonomical annotations (8) refer to one or more organisms (genus, species, strain if available) associated with the structure. If there are organism remappings (newer synonyms if an organism was reclassified/renamed after the publication, and older synonyms that identify the same organism in older publications), they are displayed in parentheses (previously named..., later renamed to...) (9). The taxonomical cross-references (10) link you to the NCBI Taxonomy database, and
(6) is a graphic representation of the structure in the SNFG format. Clicking on Show as text (7) explains the meaning of the used icons. Clicking on Show as text (7) displays the structure display in pseudo-graphic extended SweetDB format. You can visualy compare the graphic vs. pseudo-graphic representations of the sample structure in the figure on the right. CSDB linear code is not used for visualization; it is given for reference.
Taxonomical annotations (8) refer to one or more organisms (genus, species, strain if available) associated with the structure. If there are organism remappings (newer synonyms if an organism was reclassified/renamed after the publication, and older synonyms that identify the same organism in older publications), they are displayed in parentheses (previously named..., later renamed to...) (9). The taxonomical cross-references (10) link you to the NCBI Taxonomy database, and  icon opens the organism on the Lifemap tree of life.
Extended taxonomic and medical information (11) includes:
taxonomic domain and phylum; host organism occupied by the discussed microorganisms; organ or tissue in the associated or host organism from which the compound was extracted; life stage; disease of the host associated with the compound or microorganism. Diseases are linked with the International classification of diseases, ver. 11. Medical termes are linked to the MeSH database.
Extended bibliographic information (12) includes:
a flag identifying whether the structure was elucidated within the associated publication; www-address of the publication; bibliographic cross-references (PubMed ID, DOI, journal or chapter NLM ID); publisher company; corresponding author's email; list of authors' affiliations. Two white boxes display the publication abstract (13) and keywords (14).
Extended compound information (15) includes:
structure type (oligo, mono, repeating unit etc.); polymerization degree or molecular weight; chemical formula; location of the structure inside the article; originally published erroneous structure (to keep it searchable, if the structure was revised); aglycon information; trivial name; and compound class.
The next block stands for other publication-specific information on the structure (16):
experimental methods described in the paper; biological activity information; enzymes that releaze or process the structure; availability of synthetic, biosynthetic, genetic and conformation data in the paper; other comments (e.g. errors in this publication found elsewhere).
The related records (17) is a list of links to the CSDB records that contain the same or related structures (products of degradation, molecule parts, biochemically related, etc.) or the same publication. The links in this section include cross-references to other databases (CCSD, GlycomeDB, CAS registry, patent numbers, etc.), if available.
The NMR information includes:
temperature, solvent, chemical shift reference if not TMS (NMR conditions) (18) used in 1D NMR experiments, and NMR signal assignment tables (19) for 1H and 13C. The linkage column in these tables indicate the path to the residue from the reducing end or from the rightmost residue in the repeating unit. An experimental 13C NMR spectrum is schematically plotted beside the table (20).
If structural uncertainties lead to multiple moieties, the list of chemically distinct variants is provided in selector (21). As soon as a structure is selected, its structural formula (22)is updated. At the bottom line of this window, there are several related links:
SVG generates a vector file (SVG) with a structural formula;
MW generates a brutto formula and calculates the molecular weight;
SMILES shows a SMILES code for this structural formula, and provides a link to ChemSpider search;
Show 3D opens an applet to access a 3D structure and atom coordinates (23);
Ionize realculates the molecule with ionized proton-donor and acceptor groups (-NH3+, -COO-, etc.).
In a 3D window, the molecular model can be rotated, zoomed , panned, opened in LiteMOL (icon in the lower left corner), exported to MOL, PDB, and GLYCAM-PDB formats (links in the upper right corner), stripped of hydrogen atoms, and copied to clipboard as a CSBD Linear code. Color/Decolor switches the coloring mode: by residues in the SNFG color scheme, or by atoms. Balls/Sticks/Spheres selects the rendering mode. Oligomer link converts a polymeric structure to an oligomeric repeating unit. More operations on the atomic model are available in the applet menu (Ctrl-click).
The record is followed by three structure-related tools: converter to other glycan-encoding languages (GlycoCT, LINUCS, WURCS, GLYCAM, etc.) (24); 13C and 1H 1D/2D NMR spectra simulator (25) (click here for details); and 3D visualization and/or conformation analysis by Sweet-II engine or by AMBER/GLYCAM (26).
icon opens the organism on the Lifemap tree of life.
Extended taxonomic and medical information (11) includes:
taxonomic domain and phylum; host organism occupied by the discussed microorganisms; organ or tissue in the associated or host organism from which the compound was extracted; life stage; disease of the host associated with the compound or microorganism. Diseases are linked with the International classification of diseases, ver. 11. Medical termes are linked to the MeSH database.
Extended bibliographic information (12) includes:
a flag identifying whether the structure was elucidated within the associated publication; www-address of the publication; bibliographic cross-references (PubMed ID, DOI, journal or chapter NLM ID); publisher company; corresponding author's email; list of authors' affiliations. Two white boxes display the publication abstract (13) and keywords (14).
Extended compound information (15) includes:
structure type (oligo, mono, repeating unit etc.); polymerization degree or molecular weight; chemical formula; location of the structure inside the article; originally published erroneous structure (to keep it searchable, if the structure was revised); aglycon information; trivial name; and compound class.
The next block stands for other publication-specific information on the structure (16):
experimental methods described in the paper; biological activity information; enzymes that releaze or process the structure; availability of synthetic, biosynthetic, genetic and conformation data in the paper; other comments (e.g. errors in this publication found elsewhere).
The related records (17) is a list of links to the CSDB records that contain the same or related structures (products of degradation, molecule parts, biochemically related, etc.) or the same publication. The links in this section include cross-references to other databases (CCSD, GlycomeDB, CAS registry, patent numbers, etc.), if available.
The NMR information includes:
temperature, solvent, chemical shift reference if not TMS (NMR conditions) (18) used in 1D NMR experiments, and NMR signal assignment tables (19) for 1H and 13C. The linkage column in these tables indicate the path to the residue from the reducing end or from the rightmost residue in the repeating unit. An experimental 13C NMR spectrum is schematically plotted beside the table (20).
If structural uncertainties lead to multiple moieties, the list of chemically distinct variants is provided in selector (21). As soon as a structure is selected, its structural formula (22)is updated. At the bottom line of this window, there are several related links:
SVG generates a vector file (SVG) with a structural formula;
MW generates a brutto formula and calculates the molecular weight;
SMILES shows a SMILES code for this structural formula, and provides a link to ChemSpider search;
Show 3D opens an applet to access a 3D structure and atom coordinates (23);
Ionize realculates the molecule with ionized proton-donor and acceptor groups (-NH3+, -COO-, etc.).
In a 3D window, the molecular model can be rotated, zoomed , panned, opened in LiteMOL (icon in the lower left corner), exported to MOL, PDB, and GLYCAM-PDB formats (links in the upper right corner), stripped of hydrogen atoms, and copied to clipboard as a CSBD Linear code. Color/Decolor switches the coloring mode: by residues in the SNFG color scheme, or by atoms. Balls/Sticks/Spheres selects the rendering mode. Oligomer link converts a polymeric structure to an oligomeric repeating unit. More operations on the atomic model are available in the applet menu (Ctrl-click).
The record is followed by three structure-related tools: converter to other glycan-encoding languages (GlycoCT, LINUCS, WURCS, GLYCAM, etc.) (24); 13C and 1H 1D/2D NMR spectra simulator (25) (click here for details); and 3D visualization and/or conformation analysis by Sweet-II engine or by AMBER/GLYCAM (26).
Home